
Missing Heritability: Much More Than You Wanted To Know
...

The Story So Far
The mid-20th century was the golden age of nurture. Psychoanalysis, behaviorism, and the spirit of the ‘60s convinced most experts that parents, peers, and propaganda were the most important causes of adult personality.
Starting in the 1970s, the pendulum swung the other way. Twin studies shocked the world by demonstrating that most behavioral traits - including socially relevant traits like IQ - were substantially genetic. Typical estimates for adult IQ found it was about 60% genetic, 40% unpredictable, and barely related at all to parenting or family environment.
By the early 2000s, genetic science reached a point where scientists could start pinpointing the particular genes behind any given trait. Early candidate gene studies, which hoped to find single genes with substantial contributions to IQ, depression, or crime, mostly failed. They were replaced with genome wide association studies, which accepted that most interesting traits were polygenic - controlled by hundreds or thousands of genes - and trawled the whole genome searching for variants that might explain 0.1% or even 0.01% of the pie. The goal shifted toward polygenic scores - algorithms that accepted thousands of genes as input and spit out predictions of IQ, heart disease risk, or some other outcome of interest.
The failed candidate gene studies had sample sizes in the three or four digits. The new genome-wide studies needed five or six digits to even get started. It was prohibitively difficult for individual studies to gather so many subjects, genotype them, and test them for the outcome of interest, so work shifted to big centralized genome repositories - most of all the UK Biobank - and easy-to-measure traits. Among the easiest of all was educational attainment (EA), ie how far someone had gotten in school. Were they a high school dropout? A PhD? Somewhere in between? This correlated with all the spicy outcomes of interest people wanted to debate - IQ, wealth, social class - while being objective and easy to ask about on a survey.
Twin studies suggested that IQ was about 60% genetic, and EA about 40%. This seemed to make sense at the time - how far someone gets in school depends partly on their intelligence, but partly on fuzzier social factors like class / culture / parenting. The first genome-wide studies and polygenic scores found enough genes to explain 2%pp1 of this 40% pie. The remaining 38%, which twin studies deemed genetic but where researchers couldn’t find the genes - became known as “the missing heritability” or “the heritability gap”.
Scientists came up with two hypothesis for the gap, which have been dueling ever since:
Maybe twin studies are wrong.
Maybe there are genes we haven’t found yet
For most of the 2010s, hypothesis 2 looked pretty good. Researchers gradually gathered bigger and bigger sample sizes, and found more and more of the missing heritability. A big 2018 study increased the predictive power of known genes from 2% to 10%. An even bigger 2022 study increased it to 14%, and current state of the art is around 17%. Seems like it was sample size after all! Once the samples get big enough we’ll reach 40% and finally close the gap, right?
This post is the story of how that didn’t happen, of the people trying to rehabilitate the twin-studies-are-wrong hypothesis, and of the current status of the debate. Its most important influence/foil is Sasha Gusev, whose blog The Infintesimal introduced me to the new anti-hereditarian movement and got me to research it further, but it’s also inspired by Eric Turkheimer, Alex Young (not himself an anti-hereditarian, but his research helped ignite interest in this area), and Awais Aftab.
(while I was working on this draft, the East Hunter Substack wrote a similar post. Theirs is good and I recommend it, but I think this one adds enough that I’m publishing anyway. You can see Gusev’s response to East Hunter here)
In an interview with Aftab, Gusev explained his philosophy like so (I am excerpting heavily from a long interview and editing for flow/emphasis; completionists should read the whole thing):
For teacher-reported ADHD, the twin heritability estimate was 69% while the GWAS-based heritability estimate [ie using genome-wide association studies where researchers actually try to find the genes involved] was just 5%; with similar gaps for other behavioral traits. These are huge differences!
If we believe the twin study estimates, then this gap implies that there is a lot of causal genetic variation out there that GWAS/molecular data is not picking up. One way to think about this is that traits that are under stronger natural selection will have more of their genetic variants driven to low frequency, and thus less detectable by GWAS. So a big gap between GWAS and twins could imply that rare variants are very important due to strong selection. On the other hand, if we are skeptical of the twin study estimates, then this gap implies a substantial contribution from those environmental complexities I talked about previously. For a long time, the field of molecular genetics was operating under the assumption that the missing heritability was largely in the rare variants we had not yet measured. But a number of recent advances have started to tip the scales against that argument.
First, some of the earlier molecular heritability estimates were found to be inflated by some mix of technical issues and cultural transmission, so the amount of missing heritability actually increased.
Second, a new model was developed that could estimate total direct heritability using molecular data from mother-father-child trios, with very few model assumptions (the title literally states “… without environmental bias”; Young et al. 2018), and it too found estimates that were substantially lower than twins on average.
Third, several studies have now actually measured the influence of rare variants in various forms, and they are so far not adding up to explain as much as we would expect from twin heritability estimates.
Fourth, there is little evidence of the strong natural selection that would be needed to generate a massive trove of rare variants untagged by GWAS. I am a molecular geneticist, and this drumbeat of evidence from molecular data has convinced me that twin studies are either 2-3x inflated or estimate something fundamentally different from direct heritability.
We’ll start by looking at Gusev’s first claim: that “earlier molecular estimates” (ie polygenic scores) are significantly inflated, or at least don’t mean what we thought they meant. This won’t be directly relevant to our question - even our original number of 17% implies missing heritability2, so moving it down a bit to 5-10% or up a bit to 20% doesn’t add or subtract from the fundamental mystery. But this discussion has gotten a lot of people extremely confused, and we’ll need to deconfuse ourselves if we’re going to get any further.
Are Most Current Polygenic Scores Confounded?
A polygenic score is one possible result of a genome-wide association study. These scores are algorithms which take a person’s genes as input and return information about their traits as output. Better polygenic scores can predict a higher percent of variance in a certain trait. For example, the latest polygenic score on educational attainment can predict up to 17% of the variance in how much schooling someone completes.
Predictive power is different from causal efficacy. Consider a racist society where the government ensures that all white people get rich but all black people stay poor. In this society, the gene for lactose tolerance (which most white people have, but most black people lack) would do a great job predicting social class, but it wouldn’t cause social class3. It certainly wouldn’t be a “gene for social class” in the sense where it controls the part of your brain that helps you manage money, or where genetic engineering on this gene would make people richer.
Here are three common ways that not-directly-causal genes can show up as predicting a trait:
Population stratification: genes are linked to culture, and culture determines the trait, as in the racism-lactose example above. Many studies naturally mitigate this concern by using the UK Biobank of mostly white British samples, and by correcting for “principal components” that correspond to ancestry (and there are other, even more complicated ways to correct for this). But ancestry variation is fractal; no matter how uniform your sample, there will still be micro-differences you didn’t consider. For example, if you’re analyzing the educational attainment of white British people, it’s very relevant that families with Norman surnames still outperform their Saxon peers at Oxbridge admissions 900 years after William the Conqueror. If Britons with more Norman ancestry have non-education-related genes that their Saxon peers lack, these could be mistakenly classified as genes for education or other behavioral differences between the two groups.
Assortative mating: Suppose that both height and wealth are desirable qualities in a mate. Then tall people will tend to marry rich people, and over generations, the same people will be both rich and tall. That means that even if wealth is 0% genetic, a study looking for “the gene for wealth” will be able to find genes that rich people have more often than poor people - namely, the genes for height.
Or suppose that smart people tend to marry other smart people - surely true, if only because so many couples meet at college. Then all the intelligence genes will concentrate in the same people. So any study that tries to determine how much Intelligence Gene ABC affects intelligence will get inflated4 results, because everyone with Intelligence Gene ABC will also have many other intelligence genes - if the study naively asks “How much smarter are people with Gene ABC than people without it?”, it will find they are much smarter (because it’s accidentally including part of the effects of all the other intelligence genes that travel along with it).
Parent-to-child transmission, aka “genetic nurture”: Children tend to share their parents’ genes. So if there’s a gene that causes parents to create a certain kind of childrearing environment, and that childrearing environment affects a trait, it will falsely look like a gene that directly causes the trait.
Suppose Gene XYZ causes parents to read more books to their children, and reading books to children increases their IQ. Parents with Gene XYZ will tend to read books, so their kids will get high IQ. Those kids will also (probably) inherit Gene XYZ from their parents. So people with Gene XYZ will tend to have higher IQ. If you naively study which genes increase IQ, you’ll see Gene XYZ in more smart people than dumb people, and think it’s a “gene for IQ”. This is “causal” in a certain sense, but it’s not the one we traditionally think about, and it behaves importantly differently - for example, if you genetically engineer someone to have Gene XYZ, their IQ won’t go up (although their kids’ IQs might).
How can we tell if a polygenic predictor is “direct” vs. confounded by these non-causal pathways? The most common technique is within-family comparisons: do the traditional “check if people with the gene differ on a trait from people without the gene” study, but limit its focus to (for example) sibling pairs. Suppose a couple has two children; the first child inherits Gene ABC and the second one doesn’t. If the first child is smarter than the second child, that provides some infinitesimal evidence that Gene ABC is a gene for intelligence. Repeat this process over hundreds of thousands of sibling pairs, and the infinitesimal evidence can reach statistical significance. Since the family unit is a perfect natural experiment that isolates the variable of interest (genes) while holding everything else (culture and parenting) constant, within-family results are protected against stratification, assortative mating, and genetic nurture effects.
The culmination of this research program is Tan et al 2024, which finds that many polygenic predictors lose significant accuracy when retested among siblings.
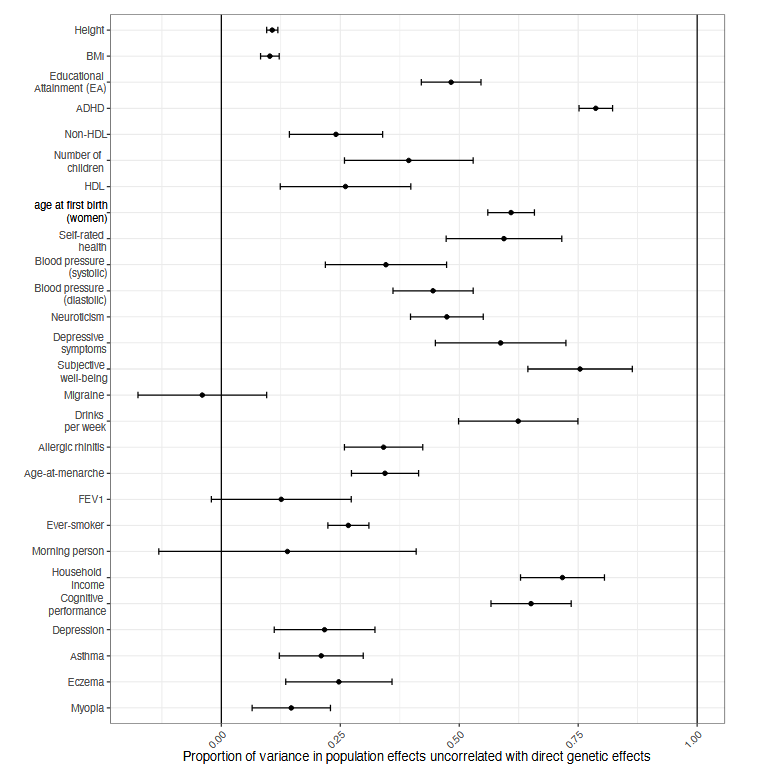
For example, educational attainment is 50% uncorrelated with direct genetic effects. You need to square this to figure out what percent is causal; when you do that, you find that the polygenic score that explained 14% of EA is only 4%pp direct genes, with the other 10%pp being nondirect5 confounders.
So yes, it seems like most polygenic scores that don’t validate within families are confounded. However unhappy we previously were that we had only found 14% of genes for EA (vs. 40% expected), we should now be much more unhappy - we really only know 4% of genes that directly cause EA.
On the other hand, you might say - so before we only knew 14%pp out of 40%. Now we only know 4%pp out of 40%. This is discouraging, but it doesn’t fundamentally change what we know about nature vs. nurture. Both 4%pp and 14%pp are less than 40% - with either number, we must be missing something or doing something wrong. Probably that’s insufficient sample size. We’ll keep working on sample size and other things, and eventually scrounge up the missing 26%pp or 36%pp or whatever of the variance, so this doesn’t change anything. All it means is that one predictive method that the average person never knew about in the first place doesn’t work as well as we thought. Who cares?
Not doctors. So far this research has only just barely begun to reach the clinic. But also, all doctors want to do is predict things (like heart attack risk). They don’t care if they use causal vs. nondirect genes. It doesn’t matter if you’re “only” at higher risk of heart attack because you’re black, or Norman, or because your parents read books to you - you still need more heart attack medication!
Polygenic embryo selection companies should care. They offer polygenic scores that can be used to select healthier or smarter embryos. If the predictors they use rely partly on variants that aren’t causal within families, their real benefits could be far lower than advertised. I talked to one of these companies, who said they’d already adjusted for these effects and expected their competitors had too - the proper antidote to this problem, sibling controls, is a natural choice when you’re literally picking between siblings.
The biggest losers are the epidemiologists. They had started using polygenic predictors as a novel randomization method; suppose, for example, you wanted to study whether smoking causes Alzheimers. If you just checked how many smokers vs. nonsmokers got Alzheimers, your result would be vulnerable to bias; maybe poor people smoke more and get more Alzheimers. But (they hoped) you might be able to check whether people with the genes for smoking get more Alzheimers. Poverty can’t make you have more or fewer genes! This was a neat idea, but if the polygenic predictors are wrong about which genes cause smoking and what effect size they have, then the less careful among these results will need to be re-examined.
But the reason I spent so much time on the subject here is that this has confused a lot of people into thinking heritability itself was confounded and is actually just 4%. When I read my first few blog posts on these findings, I came away thinking they were claiming to have discredited twin studies and heritability. And although I take partial ownership of my own poor reading comprehension, I maintain that the way that the new anti-hereditarians discuss this is pretty bad. For example, Turkheimer’s treatment of the Tan study above is called Is Tan Et Al The End Of Social Science Genomics?, and includes passages like:
The median [direct genomic effect] heritability for behavioral phenotypes is .048. Let that sink in for a second. How different would the modern history of behavior genetics be if back in the 80s one study after another had shown that the heritability of behavior was around .05? When Arthur Jensen wrote about IQ, he usually used a figure of .8 for the heritability of intelligence. I know that the relationship between twin heritabilities and SNP heritabilities is complicated, and in fact the DGE heritability of ability is one of the higher ones, at .2336. But still, it seems to me that the appropriate conclusion from these results is that among people who don’t have an identical twin, genomic information is a statistically non-zero but all in all relatively minor contributor to behavioral differences.
And comments included things like:
I don’t know if [this study] is the end of social science genomics, but it should certainly be the end of attributing significant genetic influence to behavioral traits (despite the recent scientist-generated cartoons touting genes for “income”).
And:
There's no doubt that this reported findings have dealt a fatal blow to my conviction that behavioral traits are pre-eminently heritable…This is a remarkable example of an objective statistical fact mercilessly crushing the more subjective experiential sense of "A looks and acts more like B than C because A and B have the same parents." This subjective evidence is almost unshakable and universal in its application as a tried and tested psychosocial heuristic. And yet, here we are.
Turkheimer is either misstating the relationship between polygenic scores and narrow-sense heritability, or at least egging on some very confused people who are doing that, and the dynamic was bad enough that I got confused myself for a while.
But even more confusing, the new anti-hereditarians actually are saying that lots of behavioral traits have very low heritability! But this point requires different arguments, only tangentially related to these. So let’s move on to…
Is Heritability Genuinely Low? (Part 1: GWAS & GREML)
In the mid 2010s, when genome-wide association studies (GWAS) based polygenic predictors were getting better every year, it was easy to hope they might reach 40% and close the “missing heritability”. But since then, progress has stalled. The second-to-last tripling of sample size, from 300K to 1M between 2016 - 2018, increased predictive power from 6% → 12%. The last tripling, from 1M to 3M between 2018 - 2022, only increased predictive power from 12% → 14%. If you graph sample size vs. predictive power, it looks like there's an asymptote between 15 - 20% or so.
(of which - remember - only 5% is directly causal!)
Worse, a mid-2010s technique called GREML allowed researchers to estimate the percent of variance in a trait that comes from the sorts of common genes studied in GWAS, without having to identify the genes involved. A 2016 GREML paper suggested that the maximum share of variance that GWASs of educational attainment could ever discover was about 21% (again, compared to 40% predicted genetic from twin studies). Since unavoidable methodological issues will prevent GWASs from reaching the literal maximum possible, this agrees with the evidence suggesting an asymptote between 15 - 20%.
So either twin studies are wrong and traits are less heritable than believed, or the heritability must lie somewhere other than the common genes identifiable by GWAS.
What about rare genes?
GWASs focus on genetic variation common enough to be worth including in a basic genetic test. Most of this is single nucleotide polymorphisms (“SNPs”). A single nucleotide is one letter of DNA - for example, a C or a G. Polymorphisms are genes that commonly vary in humans - sometimes across races (for example, some humans have a gene for light skin, and other humans have a gene for dark skin), and other times within races (for example, some white people have a gene that makes cilantro taste like soap, and others don’t). So SNPs are single-letter spots in DNA where different people often have different letters. How often? Some people say 1%, but the more practical definition is “often enough that someone has noticed and added it to the test panel”. There are three billion letters in the genome, of which only a few million are commonly-tested SNPs.
But these SNP studies have limited7 ability to measure personal mutations and rare variants. Sometimes your parents’ egg and sperm cells mess up copying a nucleotide of DNA, and you get a mutation that isn’t inherited from your ethnic group or even from your subgroup/family line - it’s just some idiosyncratic DNA change that you might be the first person in history to have. Since scientists have never seen this mutation before, they don’t know about it and can’t test for it without doing something more expensive than a simple SNP screen.
And SNP studies have limited ability to detect anything more complicated than a single letter changing to another single letter. But some mutations are more complicated structural variants. For example, some bits of DNA get stuck on repeat - one person might have GATGAT, another person might have GATGATGATGAT, and a third person might have fifty GATs in a row. Other bits come out backwards. Sometimes a whole chunk of DNA goes missing, or moves to the wrong place. Occasionally a gene reads The Selfish Gene by Richard Dawkins, takes it too seriously, and evolves some ridiculous trick for spamming itself all over the genome.
So if even the best molecular studies seem to be asymptoting around 15-20% of variance in educational attainment, but twin studies suggest it’s 40% genetic, might rare variants and structural variants make up the missing 20-25%pp?
This remains a topic of bitter disagreement.
On the one side, hereditarians bring up a Darwinian argument: imagine a genetic engineer who hopes to find the genes for educational attainment and edit them to make everyone smart and successful. She looks harder and harder, becoming more and more exasperated as they fail to materialize. Finally, she realizes she’s been scooped: evolution has been working on the same project, and has a 100,000 year head start. In the context of intense, recent selection for intelligence, we should expect evolution to have already found (and eliminated) the most straightforward, easy-to-find genes for low intelligence. Therefore, everything left should be convoluted or hidden or impossible to work with. So although this requires a sort of god-of-the-gaps argument - where we keep pushing heritability into whatever genes are too weird for existing techniques to detect - there are some reasons to think God really is in the gaps here. And a 2017 paper uses some clever techniques to estimate the share of intelligence variation lurking in hard-to-measure genes and finds it’s more than half: “By capturing these additional genetic effects, our models closely approximate the heritability estimates from twin studies for intelligence and education.” (see also Wainschtein 2022, Sidorenko 2024)
The anti-hereditarians disagree. They cite papers like Zeng which measure the strength of selection on intelligence and suggest that it’s too weak to concentrate so much of the variation in rare genes8. And Sasha Gusev mentions Weiner 2023, which finds that in fact rare variants “explain 1.3% (SE = 0.03%) of phenotypic variance on average – much less than common variants” (other experts say that burden heritability only captures some rare variants and is not the right tool for this problem).
But it may not even matter, because another set of findings suggests that heritability is genuinely low even when the rare variants are counted.
Is Heritability Genuinely Low? (Part 2: Sib-Regression and RDR)
Two newer methods, Sib-Regression and RDR, ask: using what we know from genetic studies, how much genetic variation do we think exists, total, across both common and rare genes?
On average siblings share 50% of genes. But there’s a little randomness in meiosis, so some siblings might share 40% and others might share 60%. The more genetic influence on a trait, the more similar sibling pairs who share 60% of their genes will be, compared to sibling pairs who only share 40% of their genes. Since 60%-gene siblings and 40%-gene siblings are both equally part of the same family, you can use these numbers to calculate heritability unconfounded by a range of family factors. This is Sib-Regression. If you do a more complicated statistical process to extend the same idea to relatives other than siblings, it’s relatedness disequilibrium regression or RDR.
GWAS asks: Looking at common easy-to-study genes, how much variation in a trait have we explained right now? GREML asks: looking at common easy-to-study genes, how much variation could we ever explain? But sib-regression and RDR ask a question more like twin studies: considering all genes, whether common / rare / easy-to-study / hard-to-study, how much variation is there total? This could address the rare variant objection mentioned above. And in many ways, these techniques are better than twin studies - Sib-Regression eliminates many potential biases, and RDR eliminates even more (although it’s harder to pull off, requiring more genetic information and computational resources).
These techniques are new and hard-to-use, and only a few published studies have applied them to the sorts of behavioral traits we’re interested in:
Young et al (2018) did Sib-Regression and RDR to genetic data from Iceland. Sib-regression found educational attainment = 40% (±15%) heritable, and RDR found 17% (±9%) heritable.
Kemper et al (2021) did Sib-Regression only to genetic data from Britain. It found educational attainment = 14% heritable. This number conflicts with the 40% from the Young paper. Why? Unclear, but it could be selection bias - Young’s Icelandic sample was representative of the country; Kemper’s British population were Biobank volunteers who tend tend to be healthier and higher-class than the population at large. Upper-class people may have restricted range in educational attainment, or different factors affecting their educational attainment compared to the overall population.
Either way, these are closer to the low estimates from GWAS and GREML (7% direct, 20% total), than to the higher estimates from twin studies (40%, generally presumed direct). And we can no longer use contributions from rare variants to paper over the difference. So what is going on?
It seems like we have to accept one of three possibilities:
Either something is wrong with twin studies.
Or something is wrong with Sib-Regression and RDR (and then we can explain away GWAS and GREML by saying they’re missing rare variants).
Or something is wrong with how we’re thinking about this topic and comparing things.
What’s Going On? (Part 1: Is Something Wrong With Twin Studies?)
Twin studies have dominated discussion of behavioral genetics for decades, so there’s a vast literature investigating their various assumptions and whether something might be wrong with them.
Here are some of the assumptions and what the research says about each. Some of these will be duplicates of the GWAS confounders above, but we’ll go through them again anyway to review how they apply to twins.
1: Parents Treat Fraternal And Identical Twins The Same: Twin studies claim that twins are a uniquely powerful genetic laboratory; both fraternal and identical twin pairs have equally concordant environments, but identical twins have more concordant genes. Therefore, the more similar identical twin pairs are relative to fraternal twin pairs, the more heritable a trait must be. But this conclusion falls apart if identical twin pairs actually have more similar environments than fraternal twin pairs do, maybe because parents (knowing their twins are identical) treat them more similarly than they would fraternal twins.
Would-be twin-study-discreditors have been trying to argue that this must be true for decades, but it’s always been a kind of quixotic battle. Remember, twin studies find many behavioral traits like IQ are >60% heritable, so you would need to prove not only that parents treat identical twin pairs differently from fraternal, but that this was an overwhelming effect. Parents of identical twins would have to obsessively expose them to the exact same stimuli in the exact same order; parents of fraternal twins would have to send one to the Gifted Advanced Placement Acceleration program while locking the other in a box and force-feeding them lead pellets. Common sense tells us there are no such differences, and studies confirm this: when parents are wrong about their twins’ status (eg they have fraternal twins, but falsely think they’re identical, or vice versa) their trait similarity matches their real status, rather than the incorrect status that determined how their parents treat them; parental treatment explains less than 1% of why identical twin pairs are more concordant (2, 3, 4). See also Felson 2013, which tries to measure environmental similarity and adjust for it, with minimal effects.

2: Fraternal And Identical Twins Have Equally Concordant Uterine Environments: Fraternal twins have different sacs in the uterus and use different placentas. Most identical twins share a placenta, and some share an amniotic sac. If trait similarity is caused by sharing a placenta or sac (maybe because the placenta is defective, the fetal brain is starved of nutrients, and so the person has a lower IQ when they grow up), twin studies would falsely read this identical-fraternal difference as genetic.
Luckily this is easy to study; not all identical twins share a placenta or sac, so you can cleanly separate the effect of uterine environment from genetics. If you measure enough traits, you can find small deviations in some, but it’s not clear whether this is just multiple testing, and in any case the deviations are small. The best studies suggest this chips off somewhere between 0 - 3% from heritability estimates9.
3: There is little assortative mating: We discussed this one above in the earlier section on GWAS - smart/pretty/kind/whatever people tend to marry other smart/pretty/kind/whatever people. Why would this bias twin study results? Identical twins share 100% of their genes. Fraternal twins ought to share 50% of their genes - but they get half their genes from their mother, and half from their father. In the degenerate case where the mother and father have exactly the same genes (“would you have sex with your clone?”) even fraternal twins will be extremely similar (although not quite identical, since they’ll get different alleles from each clone). In the more plausible case where mothers and fathers are just a little more alike than chance (eg because smart people tend to marry other smart people), fraternal twins will share a genetic tendency towards a trait somewhat more than their 50% shared genes suggest. Since this makes fraternal twin pairs more (genetically) like identical twin pairs, and twin studies assess heritability as the difference in fraternal-identical-twin-pair concordance, this bias would make twin studies underestimate heritability. But this is the opposite of what you would need to “discredit” twin studies - if this bias is true, then everything is more genetic than twin studies think. And unlike the previous two biases, this one seems real and important, so much so that when you adjust for it, the heritability of educational attainment rises from ~40% to ~50%.
I’m only mentioning this one here because some anti-hereditarians argue that you can’t trust twin studies because of assortative mating, without mentioning that this can only bias them down.
4: Population stratification: This is often large and worth worrying about, but it applies to identical and fraternal twin pairs equally, and doesn’t bias twin study heritability estimates much (though it might shift the balance between shared and non-shared environment). See eg the sentence around footnote 30 here.
5: Non-additive / “interaction” effects:
These are theoretically interesting, but all research thus far has found they are minimal (1, 2). Some experts think this may miss rarer or harder-to-find interactions; we’ll return to this later.
6: “Genetic nurture”, parent-to-child
Mentioned above: if there is a gene for reading books to kids, and reading books raises IQ, it will look like a “gene for IQ”.
This isn’t as relevant to twin study estimates of heritability, since both identical twins and fraternal twins are equally related to their parents, and any trait caused by genetic nurture wouldn’t differ between them (and therefore would not falsely appear heritable in this design). Rather, they would appear as shared environment.
7: “Genetic nurture”, sibling-to-sibling
That is, suppose your sibling’s traits influence your own development. For example, suppose your sibling has a gene that makes them sabotage your schoolwork, causing you to fail and drop out of school early. An identical twin would share this gene with their sibling more often than a fraternal twin, making it look like a “gene for doing badly at school” (since the people who have it do worse at school than those who don’t).
Why are we even talking about this? Do we really think it’s a big part of the variance in behavioral traits? Challenging twin study heritability estimates through this route requires inhabiting a weird no-man’s-land where otherwise-invisible genetic and environmental pathways suddenly flare up when you say the magic words “it was done by a sibling”. For example, this requires a strong effect of shared environment - that is, your educational attainment has to depend on whether you’re being sabotaged or not. But in general, shared environmental effects are weak. And it requires a strong effect of genes - that is, this mechanism only works if your sibling’s tendency to sabotage you is highly genetically determined. But we’re deploying this claim to deny that traits like IQ or educational attainment are highly genetically determined. So to get much out of this, the tendency to sabotage siblings would have to be more genetic than other behavioral traits!
The reason this convoluted possibility gets brought up so often is that, unlike the more plausible parent-to-child genetic nurture, twin studies can’t rule it out. So if you really want to deny twin studies, this is one of your best bets. But when investigated, this has effects indistinguishable from zero.
I’ve been a bit mean in this whole section, because people really like to dismiss twin studies as “Oh, don’t you know, those depend on assumptions, I bet you never considered that assumptions might be wrong”, and then Gish Gallop you with different assumptions until you give up. But scientists have actually done a lot of really good work checking the assumptions and they mostly hold.
An alternative way of validating twin studies (brought up by Noah Carl in this article) is to check them against their close cousins, adoption studies and pedigree studies.
Pedigree studies investigate large family trees, and check how trait similarity decreases with genetic distance. They avoid twin specific biases (like different treatment of fraternal vs. identical twin pairs, or different prenatal environments), while adding others like assortative mating. Here are the heritabilities of IQ and EA found in pedigree studies10 (see footnote for sources and caveats, and see also here and here for somewhat similar designs):

Adoption studies investigate whether adoptees’ traits are more correlated with their adoptive or biological parents. They avoid a large swathe of biases, at the risk of introducing new adoption-related biases of their own (like the possibility that agencies deliberately place adoptive children with parents who are culturally or behaviorally similar, or the possibility that adoptees were adopted late enough to still get some shared environment from their biological parents). Here are the findings of some of the largest and best11:

Both straightforwardly confirmed the larger heritability numbers found in twin studies.
I would add the evidence from some less formal “adoption studies”12. During residency, I spent a few months working in a child psychiatric hospital for the worst of the worst - kids who committed murder or rape or something before age 18. Many of these children had similar stories: they were taken from their parents just after birth because the parents were criminals/drug addicts/in jail/abusing them. Then they were adopted out to some extremely nice Christian family whose church told them that God wanted them to help poor little children in need. Then they promptly proceeded to commit crime / get addicted to drugs / go to jail / abuse people, all while those families’ biological children were goody-goodies who never got so much as a school detention. When I met with the families, they would always be surprised that things had gone so badly, insisting that they’d raised them exactly like their own son/daughter and taught them good Christian morals. I had to resist the urge to shove a pile of twin studies in their face. This has left me convinced that behavioral traits are highly heritable to a level that it would be hard for any study to contradict.

But I don’t think studies do contradict this. Given the degree to which their assumptions have been validated, and the level of confirmation from pedigree and adoption studies, I think they have earned a presumption of accuracy. Doubting the twin studies doesn’t seem like a promising route to reconciling the twin-vs-Sib-Regression/RDR discrepancy.
What’s Going On? (Part 2: Is Something Wrong With Sib-Regression And RDR?)
Sib-Regression is a clever way of avoiding most biases. Its independent variable - the degree to which some sibling pairs end up with slightly more shared genes than others - is even more random and exogenous than the difference between fraternal and identical twins. It can sometimes have biases related to assortative mating (which would falsely push heritability down), but otherwise it’s pretty good. RDR has many of the same advantages, and allows more diverse relationships and so larger sample sizes. It’s hard to think of ways these methods could be wildly off.
There is one caveat: although RDR includes most of the rare and structural variants missed by GWAS, in theory it can miss certain ultra-rare variants which are so uncommon that they aren’t shared between some of the relative pairs used in RDR. De novo variants that occurred during the subject’s own conception would be in this category, if the subject didn’t have children or didn’t pass on that gene13. This seems like a pretty small subcategory of genetic variation, and I wouldn’t normally expect that much of importance to be hiding here, but maybe it’s more important than it seems.
RDR also doesn’t include much variance caused by statistical interactions between genes. Although we said above that these are usually found to be insignificant, they might be more important in a trait like intelligence that has been under recent evolutionary selection that lops off easily-detectable sources of variance and leaves only the weird obscure ones behind. There’s limited ability for classical Mendelian dominance to affect common variants, but more complicated genetic interactions might still prove important.
Overall these are strong methods, and their failure to converge is troubling. If forced to explain them away, we might tell a story like:
So far, there is only one RDR study and a few Sib-Regression studies, so we should wait for more data before updating too hard.
Maybe ultra-rare variants are more important than we thought.
Maybe gene x gene interactions, especially epistasis, are more important than we thought.
There’s some (weak) evidence for the latter two claims: Sib-Regression, unlike RDR, includes results from certain types of ultra-rare variants and non-additive effects. In the Iceland study, Sib-Regression found EA heritability of 40% (similar to twin studies), and RDR found 17% (much less than twin studies). Maybe these make Sib-Regression better at estimating the sort of broad heritability investigated in twin studies?
What’s Going On? (Part 3: Is Educational Attainment Just Weird?)
Above, we said that there were only two published peer-reviewed studies using Sib-Regression and RDR to estimate heritability of behavioral traits.
But Markel et al (2025), a not-yet-peer-reviewed pre-print from GMU (why is it always GMU?) complicates things further. It looks at genetic data from six different countries/studies to estimate heritability of IQ and EA.
Using Sib-Regression, they find educational attainment heritability of only 8% (±9%)14, and cognitive performance (~IQ) heritability of 75% (±20%)!
Markel’s 8% for EA is very different from Young’s Icelandic estimate of 40% - is this bad? Not necessarily - as with Kemper, these studies might have different levels of selection bias. Or the countries where they take place might have different levels of educational mobility.
But also, this is the first Sib-Regression study to investigate IQ - all the others had only done EA. They replicate (and even go beyond) the twin studies’ high IQ number, while continuing to get low heritability for EA. This suggests our previous assumption - that EA was usually a decent proxy for IQ - might be totally off.
This doesn’t directly solve any of our problems - the twin study estimates for EA and the Sib-Regression estimates are still worryingly different. But it slightly bounds the damage. It suggests that the twin study estimates for IQ are ~correct, potentially meaning that whatever’s going on is some kind of EA-specific confounder.
We know that EA is a pretty unusual trait, with high assortative mating, high shared environmental component, and high potential for genetic nurture / dynastic effects. We saw above that there are theoretical reasons not to expect these to bias twin studies upward or Sib-Regression downward. But maybe it did that anyway, despite the theoretical reasons.
Stepping back, maybe educational attainment is full of landmines. Plenty of political and economic factors affect the degree to which your genes vs. your culture determine how far you go in school. Suppose a country passes a feel-good policy that high schools have to try to graduate all students, even ones who fail algebra. That changes the heritability of EA! Or suppose that scholarships become easier/harder to get, making rich people less/more likely to go to college relative to poor people. That changes the heritability of EA! Or suppose that the economy changes and jobs requiring PhDs are less/more lucrative than before - now ambitious people are less/more likely to pursue PhDs relative to people doing it for the love of academia, and that changes the heritability of EA! Finally, suppose some study enrolls mostly rich/well-educated people, and some other study enrolls proportionally across the population. That artificially restricts range and . . . changes the heritability of EA!
So two potential takeaways from this preprint are:
EA is a weird trait with a high shared environmental component, and might not be a good flagship trait to use for discussing heritability more generally.
Heritability of EA might genuinely vary a lot among different countries, populations, and eras, so maybe we should be less concerned when studies give weird results.
What’s Going On? (Part 4: Is Everything Weird?)
On the other hand, here are some boring medical/biological traits. Graph is from here, but the ultimate data source is the same Iceland paper:

Many of the boring medical traits have as much “missing heritability” as educational attainment. For example, creatinine is a measure of kidney function; although twin studies find it’s about 55% heritable, Sib-Regression and RDR find less than half that.
But here there are limited opportunities for confounders. Nobody assortative-mates on kidney function. It’s hard to see how family members could push other family members to have better or worse kidney function. Identical twins don’t have more similar kidney function environments than fraternal twins.
Could there be some remaining possibility for confounding? Maybe there’s a gene for teaching your kid to have a good diet, and good diet causes better kidney function? Or maybe the measurements for creatinine were really bad during the Sib-Regression study (but apparently better during the twin studies?)
I don’t really know what’s going on here.
What’s Going On (Part 5: No, Seriously, What’s Going On?)
So how heritable are complex traits, and why can’t different methods agree on this?
I think the twin / pedigree / adoption estimates are mostly right. They are strong designs, their assumptions are well-validated, and they all converge on similar results. They also pass sanity checks and common sense observation.
Although polygenic scores, GWAS, GREML, RDR, and Sib-Regression are also strong designs, they’re newer, have less agreement among themselves, and have more correlated error modes in their potential to miss rarer variants and interactions. Although it’s hard to figure out a story of exactly what’s going on with these rarer variants and interactions, there seems to be some evidence that they exist (again, see 1, 2, 3)15, and it seems easier to doubt this new and fuzzy area than the strong and simple conclusions from twin / pedigree / adoption work.
In this model, polygenic scores, GWAS, and GREML could straightforwardly fail to pick up rare variants and interactions. The conclusions of RDR and Sib-Regression are harder to explain, but most of these anomalies are in educational attainment in particular - which is such a cursed construct, and so variable from sample to sample, that perhaps we can put it aside and focus on more stable traits. Otherwise, we can take solace in these methods’ failure to stay consistent even among themselves, which makes their inconsistency with twin studies somewhat less jarring.
What mysteries remain? The parts that still bother me are:
Why did the Iceland study find significantly lower numbers for Sib-Regression/RDR than twin studies for almost every trait? (hilariously, not for educational attainment with Sib-Regression this time, although I suspect this is just the big margin of error and the real number is commensurate with the other traits studied)
Why are twin studies so consistent in finding highish heritability for educational attainment, but other methodologies (eg Sib-Regression) so variable (even after their margin of error is taken into account)? Either EA is too complicated and variable to measure properly, or it isn’t - right?
If nonadditive interactions are so important, why have existing studies had such a hard time detecting them?
Can we flesh out the evolutionary argument and come to agreement on how many rare and ultra-rare variants we should expect given the level of selection pressure experienced during human evolution?
Can we bound the degree to which ultra-rare variants can drive a wedge between twin and RDR estimates? If the observed difference isn’t within the bound, what then?
Are we going to find and cash out “rare variants and interactions” soon? If we don’t, how long should we wait for genetic science to advance before changing our mind and deciding we must be missing something more fundamental?
Alex Young thinks that once we get enough whole genomes sequenced (probably soon!) we might be able to use a technique called GREML-WGS to get more definitive answers about rare variants. But other experts I talked to said that if complex interactions were a big part of the picture, this might be “computationally intractable”. On the other hand, “computationally intractable” is a relative term: with enough data, genomic language models offer the potential for improved understanding of nonlinear effects.
I’m encouraged to see increasingly good discussion of these topics on Substack, Twitter, and elsewhere. People like Sasha Gusev and Eric Turkheimer deserve credit for opening the discussion, but I would like to see a robust back-and-forth with the other side.
Thanks to everyone who helped me review this post, including Ruben Arslan, Alex Young, Damien Morris, and some other people who didn’t respond to my email asking if I had their permission to list their names publicly (if this is you, let me know and I’ll edit you in). Most of what’s valuable is theirs, and all errors are mine alone the fault of o3, which provided invaluable research assistance but also hallucinated constantly.
I’m abbreviating “two percentage points” as 2%pp. Nitpickers complain if I don’t use the “percentage points” framing, but it’s too long to spell out each time.
Geneticists distinguish between three related concepts:
Polygenic score r^2 is the degree to which our current best genetic models can predict traits. You might use this to discuss the accuracy of a genetic test or an embryo selection procedure.
Narrow sense heritability is the degree to which all normal, additive genetic variation affects traits. You might use this to discuss the effectiveness of breeding programs, or how much you expect a parent’s traits to affect their children.
Broad sense heritability is the degree to which all genetic variation, including interactions and rare mutations, affect traits. A correctly-done twin study should (modulo certain small issues) return the broad sense heritability. This is useful in resolving deep questions like “How much do genetic vs. social causes affect traits”, and acts as the limit for what we might be able to explain through some future genetic science.
If there were no missing heritability, we should mostly expect polygenic score r^2 to converge to narrow sense heritability, modulo a bunch of small biases and technical issues. I’ll be sweeping these under the rug and talking about narrow sense heritability as the limit of polygenic score r^2 in order to highlight the question of why polygenic score r^2 seems to be asymptoting at a level below the narrow-sense heritability.
Why is this example about a gene for lactose tolerance, rather than simply a gene for black skin? Because in this society, the gene for black skin would in some sense “cause” the poverty - it would just be a less direct form of causation, through “gene-environment interaction”. Causal language in genetics gets tough quickly - what is a “confounder” for one purpose may well be a “cause” for another.
"Inflated” if you were hoping to find the direct heritability. If you just want predictive power, this might be fine.
I’m using the awkward term “nondirect” instead of the more common “indirect” at the advice of an expert who advised that “indirect genetic effect” already has a specific meaning (similar to “genetic nurture” described elsewhere in this piece) and should not be used for things that merely fail to be direct.
Sic; the correct number is 18.8%
Not zero, because some rare variants are linked to SNPs; that is, everyone with a certain rare variant also has a certain set of SNPs, and so in practice measuring the SNPs will include the effect of the rare variant.
I don’t know how you square that with Reich’s study indeed finding large recent selection for intelligence in ancient European DNA
See this Cremieux tweet thread for some apparently paradoxical - though ultimately inconsequential - effects of uterine-environment-sharing
Sources:
Generation Scotland: https://www.sciencedirect.com/science/article/pii/S0160289614000178?via%3Dihub . See also https://www.nature.com/articles/s41380-017-0005-1.pdf, page 2353, "The genetic results . . . are similar to the heritability estimates derived using the traditional pedigree study design in the same data set, which found a heritability estimate of 54% for g and 41% for education."
Swedish National Sample: https://www.nature.com/articles/s41380-022-01500-2/tables/2, Table 2B, first column, second and third rows.
I made this table with the help of the o3 AI. It gave me a longer list of seven studies, but admitted that many of them didn’t list heritabilities and it was calculating them itself based on other statistics that were reported. In some cases, I was able to replicate its calculations; in others, it seemed to be hallucinating the relevant numbers, or had calculations complicated enough that I couldn’t prove that it wasn’t. I excluded two studies that o3 said had good data, but where it couldn’t explain its sources well enough to satisfy me. These were Kendler et al 2015 (o3 claimed heritability of 0.5 - 0.6) and Capron & Duyme 1989 (o3 claimed heritability of 0.4).
Here are sources for the remaining:
Texas adoption: https://gwern.net/doc/genetics/heritable/adoption/2021-loehlin.pdf , Table 6 . Heritability = 2*correlations. The first row of the table lists three parent-child IQ correlations (IQBM): 0.3, 0.34, and 0.35. I took the median, 0.34, and doubled it to 0.68.
Colorado adoption: Same paper as above, same methodology as above, Table 7.
Minnesota transracial: https://sci-hub.st/https://www.sciencedirect.com/science/article/abs/pii/016028969390018Z, page 547, "adoptive fathers' and mothers' correlations with their biological offspring at Time 1 were .25 and .40; with their trans-racially adopted children, .08 and .14 (h 2 = .34 ± .29 and .52 --- .26, respectively). At Time 2, the corresponding correlations were .13 and .45 for biological children; .21 and .21 for adoptees (h 2 = - . 16 ± .29 and .48 ± .25, respectively). o3 advised me to take the maternal correlation since there were too few fathers to give meaningful results (as indicated by the negative heritability at time 2). Because this required a design choice (throwing out the paternal data) I’ve listed it in gray as less trustworthy than some of the other estimates. Reversing this design choice would lower the estimate.
Sibling Interaction And Behavior: https://pmc.ncbi.nlm.nih.gov/articles/PMC8513766/, see second-to-last sentence of the abstract.
Swedish Adoption Twin Study Of Aging: https://sci-hub.st/https://doi.org/10.1111/j.1467-9280.1992.tb00045.x , see Table 3, first principal component says 80%. https://sci-hub.st/10.1126/science.276.5318.1560, abstract says 62%. These are different subsets of the same Swedish study; I averaged them together and said 71%. This is extremely unprincipled - they had different sample sizes and were measuring different things - but I also didn't want to list the same study twice.
Observed adult heritabilities of IQ range from 0.42 to 0.71, with an average of 0.57. This is a wide range, but some of the variability is explicable. The SIBS study, which found the lowest estimate, used an unusually unreliable IQ test; if you correct for this, heritability increases to about 0.6 to 0.7. SATSA, which found the highest estimate, was looking at older people (age 65 - 80), and heritability of IQ increases with age. I think the adoption literature looks compatible with heritability of IQ going from the 40s in adolescence to the 60s in adulthood to potentially as high as 70-80% by age 65 (the 65 year olds in SATSA found 80%; heritability declined after 65, maybe because of non-heritable dementias).
And here’s a less formal “twin study” - on average, twins have the same IQ and educational attainment as singletons. As a parent of twins myself, I’m increasingly aware how much worse parenting I do than my singleton-parent friends; it’s not just that you have half as much time per child, but that you can almost never give either of them quality time alone - the other one gets jealous and immediately interrupts. But this lower-quality parenting seems to have no measurable real-world effect!
These might or might not be a problem with sibling-based estimates, depending on how the mutation happened - some mutations can happen in pre-sperm stem cells and affect many different sperm, so multiple siblings could theoretically be affected.
When the authors correct for assortative mating, heritability of EA goes up to 9-15%, depending on how much assortative mating they assume.
I tried to create a very weak and made-up model of what I thought was going on to see whether I could get the various study types to make sense. Here is the best I could do - please don’t take this seriously:

These errors are mostly within the various studies’ margin of error. I’m very unsure that I’m parsing the difference between common, rare, and ultra-rare right